AlphaFold: Artificial Intelligence Platform Towards Calculation of Protein Structures
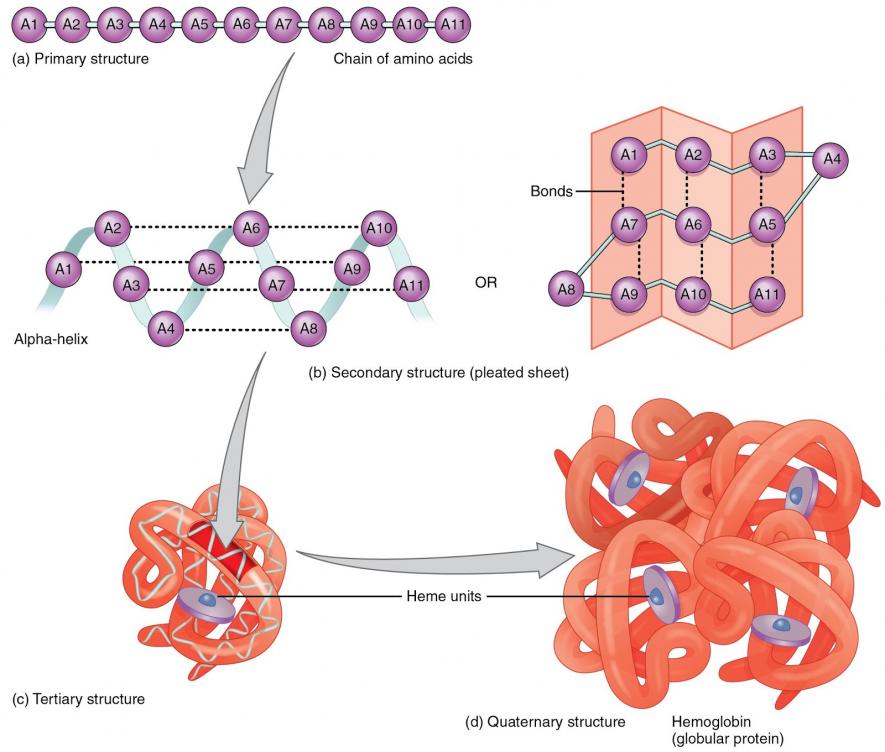
Image Courtesy: Wikimedia Commons
Proteins are primary in the functioning of any life forms. A protein molecule, in its functional state exists in a complex three-dimensional structure. How a protein perform its functions depends overwhelmingly on the three-dimensional structure that it folds upon. A protein molecule derives its functional structure from the linear sequence of amino acids, the building blocks of proteins.
A significant question in biology has been, how does a protein take the three-dimensional structure based upon the amino acid sequence? More importantly, the question that has been disturbing biologists is that whether one can determine the exact functional structure that a protein molecule shapes up from the information of the amino acid sequence?
The latest research in this field has used artificial intelligence (AI) that could predict the protein structure given its linear amino acid sequence. DeepMind, an UK based company has declared to have achieved protein structure prediction with accuracy more than the already available computer tools.
Before we understand the latest achievement, it is important to consider the Levinthal’s paradox, which emerged as a thought experiment almost half a century back. Cyrus Levinthal in 1969 calculated that a manual calculation, using however sophisticated tools, of a protein’s structure from the amino acid sequence would take an astronomical time. Whereas, in natural condition a protein folds to its three-dimensional structure even within a fraction of a time. Since then, biologists have been striving to develop a way to determine the protein structure using various tools, as sophisticated as X-ray crystallography and the NMR (Nuclear Magnetic Resonance).
However, even in these techniques, predicting the structure of a protein requires painstaking work worth months of efforts. Carrying this forward, of late, computational tools have been utilised for solving the paradox. The idea is, if someone has the mere amino acid sequence of a protein and feeds this information to a computer platform then the final structure could be predicted within hours, or even within minutes in some cases.
DeepMind Research
DeepMind has used the algorithm named AlphaFold in achieving the target. This was declared by the organisers of the CASP (Critical Assessment of Protein Structure Prediction), a biennial competition that assess the protein predicting tools in terms of their accuracy.
Commenting on AlphaFold’s ability, John Moult, a co-founder of the CASP and a structural biologist at University of Maryland said, “This is a 50-year-old problem. Never thought I’d see this in my lifetime.” Janet Thorton, the director emeritus of the European Bioinformatics Institute (EBI) commented, “What the DeepMind team has managed to achieve is fantastic and will change the future of structural biology and protein research.”
The quest to computationally predict a protein’s 3D structure based on only the linear amino acid sequence was sparked back in 1972, when Christian Affinsen, in his acceptance lecture of the Nobel prize in chemistry said that theoretically it is possible to determine a protein’s structure fully based on the amino acid sequence.
Attempts of computational prediction of protein structure had been going on since then and it took another shape in 1994, when professor John Moult and Professor Krzysztof Fidelis founded the CASP, the biennial blind assessment platform to monitor progress in establishing a state of the art system of protein structure prediction.
The CASP uses an assessment matrix called the GDT (Global Distance Test) that have a range of scores from 0 to 100. The GDT score can be thought as the percentage of amino acid residues’ distance from the correct position. Professor Moult says that a 90 GDT score is considered to be competitive in comparing the results from experimental methods like X-Ray Crystallography and NMR.
The AlphaFold has been declared to have scored a 92.4GDT in the CASP 14 results, the latest one. AlphaFold first participated in the CASP in 2018, the CASP13 with their results published in Nature.
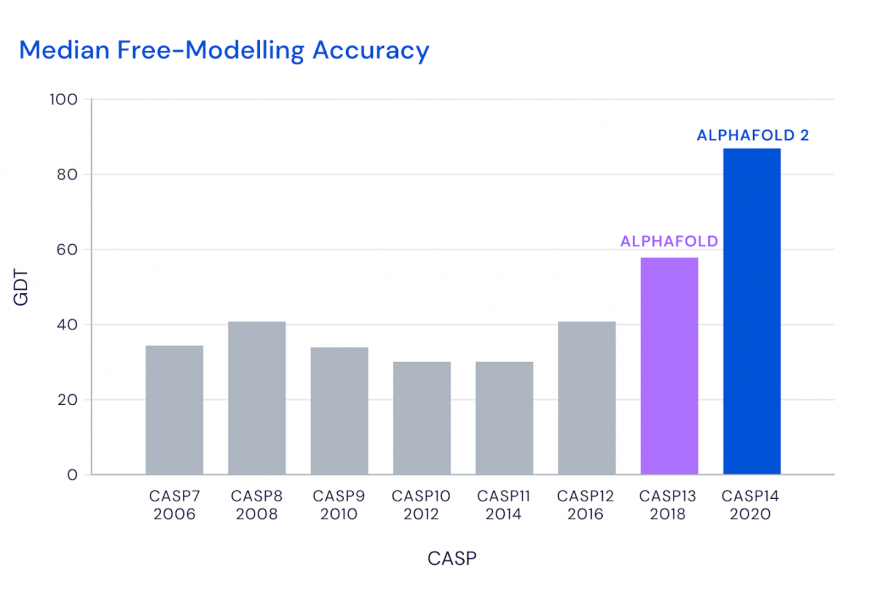
A comparative Median Accuracy Prediction of different CASP since 2006. The bar diagrams of each CASP shows the scores of the best predicting tool of that year. Image source: Deepmind.
The latest version of the AlphaFold at CASP 14 used a neural network architecture in the process known as deep learning, an AI platform. The neural network was trained with numerous other protein structures that are already available. When a novel sequence of amino acid is fed to the algorithm, it uses the knowledge that was obtained from the training that it received. The algorithm assesses the probable structure of the novel protein based upon its knowledge of other protein structures.
The platform can predict the 3D structure of a protein within days based only on the amino acid sequences.
Get the latest reports & analysis with people's perspective on Protests, movements & deep analytical videos, discussions of the current affairs in your Telegram app. Subscribe to NewsClick's Telegram channel & get Real-Time updates on stories, as they get published on our website.