Blood Disorder Finds New Cure in Gene Therapy
Gene therapy might be able to cure another disorder — beta-thalassaemia, a genetic disorder, which renders the body unable to create sufficient haemoglobin, making the affected person anaemic. The treatment for this disorder requires regular blood transfusions. But in a new study published in the the New England Journal of Medicine, carried out on 22 patients around the world, the researchers were able to almost completely eliminate the need for transfusions in the patients.
Out of the 22 patients, nine patients suffered from the most severe form of the disease. Three of them did not require any blood transfusions after the therapy. In the other thirteen, twelve patients no longer needed transfusions post therapy. And in the patients who continued to require the transfusions, the frequency was significantly reduced.
"Reducing or stopping transfusions is important for patients with thalassaemia because they are a double-edged sword- essential for survival but also responsible for life-changing, life-shortening side-effects. This means that the results are very encouraging,” said Irene Roberts, professor of paediatric haematology at Oxford University.
The team of researchers, known as LentiGlobin, published their findings in the journal earlier this week. They kept track of the patients for three and a half years, and the effects of the treatment persisted in this time. However, the long term effects of genome manipulation still need to be carefully monitored.
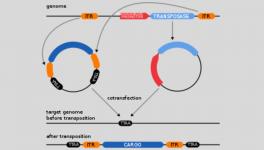
The gene therapy is carried out by first taking stem cells from the patient's bone marrow, then modifying them so that the genes can correctly carry out the functions of producing haemoglobin. This modification results in the replacement of the defective genes with the correct ones. Once modified, the stem cells are infused into the patient's bone marrow again, resulting in production of healthy blood cells.
The question is, is this approach better than the older methods of curing beta- thalassaemia? Dr. Alexis Thompson, one of the lead authors of the research and head of hematology and director of the Comprehensive Thalassemia Program at Ann & Robert H. Lurie Children's Hospital of Chicago, said, "The standard procedure for a curative option for thalassemia would be a bone marrow transplant from a brother or sister, which is not without significant risk, but more importantly, most people will not have that appropriate sibling donor.”
But with this study, the patient acts as his/her own donor. "If we can envision treating patients as if the patients were their own donors, they are not exposed to some of the risks that come from transplants when you're using someone else's stem cells,” Thomspon continued.
Almost 3,00,000 people worldwide suffer from beta-thalassaemia, and a significant number of whom reside in third world, developing countries. Gene therapy, still a relatively new technology, remains prohibitively expensive and inaccessible to most residents of these countries. Professor Douglas Higgs from Oxford University said, "A major question hanging over this approach, which is hugely expensive, is whether this procedure will ever become clinically possible in developing countries, where the majority of these disorders of haemoglobin occur.”
But even dealing with the disorder with the currently available options is expensive. Thompson said, "When you take into consideration the cost of iron chelators as well as the cost of being transfused every three to four weeks, it is also considerable,. Thalassemia care is by no means inexpensive in its current form."
As the gene therapy technique further develops, it might bring down the costs. But further research is required to properly establish the findings of this study. At the moment, the patients in the study were all 12 years or older, and were tracked for a limited duration. A follow-up phase of the study is ongoing which is looking at younger patients, as well as longer term data of the previous participants of the study.
Get the latest reports & analysis with people's perspective on Protests, movements & deep analytical videos, discussions of the current affairs in your Telegram app. Subscribe to NewsClick's Telegram channel & get Real-Time updates on stories, as they get published on our website.